
Abstract
Asplenium cuneifolium Viv. is an endangered fern species restricted to European serpentine rocks, which face a risk of being easily damaged by both natural and anthropogenic agents. Establishing a comprehensive system of effective micropropagation and long-term storage of this species is therefore desirable. Freshly collected spores of A. cuneifolium were subjected to direct storage at 5 °C and in liquid nitrogen (LN). The viability of spores stored at 5 °C decreased by 10% after 1 year, whereas storage in LN did not change the initial spore viability even after 3 years. For the initiation of the gametophyte culture, disinfected spores were sown onto half-strength Murashige and Skoog medium (½MS) supplemented with 0.06 M sucrose. Following 6 months, abundant proliferation of secondary gametophytes was achieved. For the cryopreservation of gametophytes, encapsulation-vitrification and encapsulation-dehydration, were compared. Of these two methods, encapsulation-vitrification ensured survival of gametophytes of 64.5–93%, while encapsulation-dehydration guaranteed their 100% viability. The acceleration of syngamy on secondary gametophytes was achieved using a medium with macro- and micronutrients reduced to the 1/8 of the MS free of NH4NO3 and vitamins. The syngamic sporophytes could be multiplied by somatic embryogenesis (SE) induced on etiolated stipe explants in an ½MS medium devoid of any plant growth regulators. Adding 0.3 M of sucrose to the medium almost quadrupled the efficiency of the SE. The genetic stability of gametophytes after cryopreservation, as well as sporophytes obtained from them, was confirmed by flow cytometry, amplified fragment length polymorphism, and inter-simple sequence repeat markers.
Key message
Protocols for the long-term storage of spores, proliferation, and cryopreservation of gametophytes, induction of syngamy, and multiplication of sporophytes through somatic embryogenesis have been developed for Asplenium cuneifolium Viv.
Similar content being viewed by others


Introduction
The genus Asplenium belongs to the family Aspleniaceae and consists of approximately 700 epiphytic or saxicolous species that grow in both tropical and north-temperate areas (Smith et al. 2006; Lin and Viane 2014). Asplenium cuneifolium Viv. is an example of an epipetric fern, which is characteristically restricted to serpentine rocks in Europe from the Mediterranean to Norway and from Greece to Spain (Bucharová et al. 2010). It is a rare species that merely forms small populations across its range. In Poland, it only grows in Lower Silesia, in scattered and isolated localities, mainly in the Ślęża massif (Szczęśniak 2006). According to the Polish Red Data Book of Plants, A. cuneifolium has a category ‘endangered’ (EN; Kaźmierczakowa et al. 2016), which makes it the object of interest for both in situ and ex situ conservation efforts.
Tissue culture techniques, such as micropropagation and in vitro slow growth storage, along with cryopreservation, are well-established methods of ex situ conservation. These include short-, medium-, and long-term strategies that supplement the preservation of genetic resources in the natural environment (Bunn et al. 2007; Mehltreter 2010; Pence 2014; Coelho et al. 2020). In exceptional cases, they can be even the last resort for species that face a risk of extinction. Small subpopulations of A. cuneifolium, due to their specific habitats of exposed rocks, can be easily damaged by both natural and anthropogenic agents, like persistent drought, torrential rain or uncontrolled dumping of waste in old quarries (Szczęśniak 2006). Ex situ protection of their genetic variability permits a prospective reintroduction of such subpopulations if they disappear from their natural environment (Szczęśniak 2006).
Despite Asplenium is the most species-rich genus of ferns (Xu et al. 2020), only seven species were investigated for propagation in vitro and cryopreservation. Most of the studies concerned multiplication of the ornamental epiphytic species, bird’s nest fern A. nidus L. (Higuchi and Amaki 1989; Fernandez et al. 1993; Fernández et al. 1997; Khan et al. 2008; Menéndez et al. 2011). Basic in vitro cultures of widespread maidenhair spleen wort A. trichomanes L. were established by Aldea et al. (2016). More detailed protocols for the micropropagation of gametophytes and sporophytes were published for four endangered or vulnerable epilithic ferns, namely A. adulterinum Milde, A. adiantum-nigrum L., A. cuneifolium Viv., and A. septentrionale (L.) Hoffm. (Marszał-Jagacka et al. 2005; Kromer et al. 2006; Somer et al. 2010; Marszał-Jagacka and Kromer 2011). For A. adulterinum and A. cuneifolium, conditions of the slow growth culture of prothalli were also developed, which enabled their one-year-long storage without subcultures (Kromer et al. 2006). However, longer-term storage strategies, based on cryopreservation of gametophytes, were established only for two, differing in ploidy, varieties of A. scolopendrium, European diploid A. scolopendrium var. scolopendrium (Mikuła et al. 2011) and American tetraploid A. scolopendrium var. americanum (Fernald) Kartesz & Gandhi (Pence 2015). For the latter variety, cryopreservation of clusters of sporophytic nodular tissue was also undertaken (Pence 2015). All these works focused on ensuring the viability of the plant material, without further analysis of its true-to-type status.
Most tissue cultures of Asplenium species were initiated from disinfected spores (Fernandez et al. 1993, 1997; Marszał-Jagacka et al. 2005; Kromer et al. 2006; Somer et al. 2010; Marszał-Jagacka and Kromer 2011; Mikuła et al. 2011; Pence 2015) sown onto Murashige and Skoog’s medium (MS; Murashige and Skoog 1962) of various mineral salt concentrations. In the case of A. nidus, they germinated into gametophytes within a week (Fernández et al. 1993, 1997), but for the rest of Asplenium species after about a month (Marszał-Jagacka and Kromer 2011; Pence 2015). Although culturing and multiplying Asplenium gametophytes usually posed no serious problems and could be carried out either in liquid or on solid media (Fernandez et al. 1993; Marszał-Jagacka et al. 2005; Kromer et al. 2006; Somer et al. 2010; Marszał-Jagacka and Kromer 2011), the spontaneous formation of sporophytes via syngamy often proceeded with low efficiency (Kromer et al. 2006; Marszał-Jagacka and Kromer 2011). A limited number of male gametophytes developing antheridia in culture were reported for A. adulterinum, A. cuneifolium, and A. septentrionale. This could be the reason for inefficient fertilization and development of sporophytes (Kromer et al. 2006). During the culture, the time needed for a new sporophyte to be evident varied from at least 2 months for A. adulterinum, A. septentrionale, and A. cuneifolium (Marszał-Jagacka and Kromer 2011) to 3 months for A. adiantum-nigrum (Somer et al. 2010).
Several approaches were established to accelerate and improve the efficiency of sporophyte propagation, however, most of them were based on the use of rhizomes treated with 6-benzylaminopurine (BAP; Higuchi and Amaki 1989; Fernandez et al. 1993; Somer et al. 2010) or low concentrations of 1-Naphthaleneacetic acid (NAA) and kinetin (Marszał-Jagacka and Kromer 2011). Furthermore, homogenization of sporophytes was proposed, however, in this case the formation of not only sporophytes, but also aposporous gametophytes was reported (Somer et al. 2010; Fernandez et al. 1993, 1997).
Somatic embryogenesis (SE), described for the first time for ferns in 2015 by Mikuła et al., seems to be a very attractive alternative for micropropagation of fern sporophytes (Mikuła et al. 2022b). The protocol is quick and effective, and, more importantly, it does not require the application of exogenous plant growth regulators (PGRs; Mikuła et al. 2015b), minimizing the risk of somaclonal variation. Thus, it would be desirable to implement SE for other fern species, both ornamental and those in danger of extinction. However, this process has been reported only for the tree fern, Cyathea delgadii Sternb.
The objective of this study was to develop a simple and comprehensive system for in vitro micropropagation and efficient long-term ex situ conservation of the endangered serpentine fern A. cuneifolium Viv. exploiting the phenomenon of SE and applying cryopreservation techniques.
Material and method
Plant material
Spores from one frond of A. cuneifolium were obtained from the herbarium of the Institute of Environmental Biology, University of Wroclaw, Poland.
Spore storage, disinfection, and sowing in vitro
A portion of the spores was put in airtight cryovials and placed at 5 °C (fridge) and − 196 °C (liquid nitrogen; LN). After 12, 24, and 36 months, the spores from LN were quickly re-warmed by immersing cryovials in a water bath at 38 °C for 3 min. Spores from all temperature regimes were tested for their viability by sowing in sterile distilled water (5 ml) in glass Petri dishes (Mikuła et al. 2015c).
The remaining spores were wrapped in a piece of filter paper, which was folded to form a small packet (Barnicoat et al. 2011). The packet was treated with 70% (v/v) ethanol for 30 s, then with 5% (v/v) commercial bleach (Domestos) for 20 min and finally washed 3 times with sterile distilled water, 5 min each time. The spores were blotted from the packet onto a Petri dish with growth medium (GM; ½MS macro- and micronutrients + MS vitamins + 0.06 M sucrose, pH = 5.6) and left to germinate in a tissue culture chamber at 22 ± 1 °C, under a 16/8 h photoperiod and at a light intensity of 3.5 µM m− 2 s− 1. The young primary gametophytes developing from spores were transferred separately onto the fresh GM for proliferation. Secondary gametophytes were subcultured every 6 months onto the same medium.
Cryopreservation of gametophytes
Encapsulation/vitrification
Primary gametophytes at the heart stage were encapsulated in alginate beads (Mikuła et al. 2009) and exposed to the vitrification loading solution (LS; 2 M glycerol + 0.4 M sucrose) for 20 min. Subsequently, they were treated with plant vitrification solution 3 (PVS3; Nishizawa et al. 1993) for up to 3 h, transferred to 2-ml cryovials (15 beads per cryovial) and plunged directly into LN and maintained for 3 days. The vials were then removed from LN and rapidly re-warmed in a water bath at 38 °C for 3 min. After rewarming, beads were treated with an unloading solution (US; 1.2 M sucrose) for 15 min, put on GM for 2 days in darkness, and then exposed to a 16/8 h photoperiod. Encapsulated gametophytes treated with PVS3, but not cryopreserved, were designated as controls.
Encapsulation/dehydration
In the second approach, encapsulated gametophytes were put on the preculture medium (PM; ½MS macro- and micronutrients + MS vitamins + 0.25 M sucrose + 10 µM abscisic acid; ABA, pH = 5.8) for 2 weeks. After preculture, the beads were transferred to 250 ml Erlenmeyer flasks and exposed to 3-day-long osmotic dehydration by treatment with 0.5, 0.75 and 1.0 M sucrose solution for 24 h at each concentration. Following osmotic dehydration, beads were harvested, surface-dried by air in a laminar-flow chamber at room temperature for 5 h, put into cryovials and immersed into LN as described above, and then in the same way re-warmed and cultured. Alternatively, cryopreservation via encapsulation-dehydration without preculture was tested as a way to shorten the time of the whole cryopreservation procedure.
Sporophyte culture and SE
Syngamy followed by the development of sporophytes occurred spontaneously on gametophytes cultured in GM. To accelerate this process, the reduction of the macro- and micronutrients to the 1/8 of the MS medium (1/8 MS) along with the complete withdrawal of ammonium ions and vitamins was applied (Makowski et al. 2016). Young sporophytes were transferred to jars with GM and exposed to a 16/8 h photoperiod and a light intensity of 20 µM m− 2 s− 1 for further growth, or transferred to constant darkness for etiolation and induction of SE. To regenerate primary somatic embryos, 2.5-mm-long stipe fragments were excised from the two youngest fronds and placed on induction medium (IM; ½MS macro- and micronutrients + MS vitamins + 0.03 M sucrose, pH = 5.8). They were cultured at 24 ± 1 °C in constant darkness. Secondary SE was induced on the stipes of etiolated somatic embryo-derived sporophytes that had developed 2 or 3 fronds, according to Tomiczak et al. (2018). To improve the efficiency of regeneration of secondary somatic embryos, an increase in pH up to 7.8 and a sucrose content in the basal medium up to 0.3 M were tested. The efficiency of SE was evaluated by calculating the percentage of responding explants and the average number of somatic embryos per explant after 2, 3, and 4 months of culture. Further growth of sporophytes obtained via SE proceeded in the same manner and under the same culture conditions as those obtained via syngamy.
Sporophyte acclimatization
Acclimatization was performed using sterile de-acidified peat as a substrate (pH = 6.5). Plantlets were grown in pots covered with jars under room conditions (22 ± 2 °C; RH ca. 60–80%). They were exposed to the laboratory environment for a period of 4 weeks before being transferred to greenhouse conditions. Following that, they were transferred to the outdoor field collection of the Botanical Garden.
Microscopic preparation
The method of plant material clearing in methyl salicylate (Young et al. 1979) was used to document the first developmental stages of somatic embryos. For this purpose, explants were fixed overnight in FAA (37% formaldehyde : glacial acetic acid : 70% ethanol; 1:1:18 v/v) and dehydrated in a series of ethanol solutions of increasing concentration, i.e. of 70% (2 h), 80% (2 h), 90% (2 h) and 100% (three changes, 2 h per each). After clearing for 2 h in each of the following solutions, i.e. ethanol : methyl salicylate (1:1 v/v), ethanol : methyl salicylate (1:3 v/v), and 100% methyl salicylate (two changes), explants were examined under an epifluorescence microscope (Vanox AHBT3; Olympus, Tokyo, Japan) in blue-violet light (400–440 nm). Additionally, the visualization of SE on the stipe explants was made using an environmental scanning electron microscope (ESEM; FEI QUANTA 200; 0.75 Tr, at a relative humidity of up to 100%, and reduced pressure of less than 10− 4 Pa).
Statistical analysis
Each in vitro culture experiment was repeated three times. Three Petri dishes with 15 alginate beads or 20 stipe explants per each combination of culture conditions represented each replicate. Statistica ver. 6.0 (StatSoft Polska Sp. z o.o., Poland) was used for statistical analyses. One- or two-way analysis of variance was performed. Means were compared using Tukey’s honestly significant difference (HSD) test, at the 0.05 level of probability.
Flow cytometry
Flow cytometric analysis was carried out for gametophytes cryopreserved by encapsulation-dehydration and for non-cryopreserved (control) ones as well as for sporophytes obtained from both gametophyte types. From each group, 12 individuals were chosen randomly to estimate the nuclear DNA content. Secale cereale ‘Dańkowskie’ (16.19 pg/2 C; Doležel et al. 1998) served as an internal standard. Whole gametophytes or young leaves of sporophytes of A. cuneifolium were chopped simultaneously with leaves of S. cereale using a sharp razor blade in 1.0 ml of nuclei-isolation buffer (200 mM Tris, 4 mM MgCl2·6H2O, 0.5% Triton X-100; pH = 7.5), supplemented with 50 µg ml− 1 propidium iodide (PI) and 50 µg ml− 1 RNase A. After chopping, the suspension was passed through a 50-µm mesh nylon filter. For each sample, 5000–7000 nuclei were analyzed using a CyFlow SL Green (Partec GmbH, Münster, Germany) flow cytometer, equipped with a high-grade solid-state laser with green light emission at 532 nm, as well as with SSC and FSC scatters using linear amplification. Histograms were analyzed using the FloMax software (Partec GmbH, Münster, Germany). The nuclear DNA content was calculated using the linear relationship between the ratio of the G0/G1 peak positions of Asplenium and Secale on the histogram of fluorescence intensities. Mean DNA contents calculated for each plant group were compared using Tukey’s test, at P = 0.05, using Statistica ver. 6.0 (StatSoft Polska Sp. z o.o., Poland).
DNA analysis with molecular markers
Cryopreserved and non-cryopreserved gametophytes as well as sporophytes obtained from both gametophyte types were also subjected to molecular DNA analyses. Total genomic DNA was extracted from about 100 g of gametophytic or sporophytic tissue using the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. The quantity of DNA was determined using a GeneQuant 1300 spectrophotometer (GE Healthcare), whereas its quality was assessed by electrophoresis on 1.0% agarose gel.
Amplified fragment length polymorphism (AFLP) analysis was performed as reported previously (Tomiczak et al. 2017). Briefly, DNA digestion with EcoRI and MseI restriction enzymes was followed by adaptors ligation and pre-selective PCR reaction. For selective PCR reaction, 6 primer combinations were used (Table 1). Products of amplification were analyzed on 7% polyacrylamide gels and visualized by silver staining.
For inter-simple sequence repeat (ISSR) analysis, 12 primers were used (Table 2). Amplification of DNA fragments was performed in a volume of 10 µl 1× PCR Incomplete Reaction Buffer (BIORON GmbH, Ludwigshafen, Germany), 1.25 mM MgCl2, 0.2 mM dNTPs, 1.0 µM of primer, 0.5 U SuperHotTaq DNA polymerase (BIORON GmbH, Ludwigshafen, Germany), and 15 ng of template DNA. Initial denaturation at 94 °C for 3 min, was followed by 35 cycles of 30 s denaturation at 94 °C, 30 s annealing at 53–60 °C (depending on the primer) and 90 s extension at 72 °C, ending in 10 min final extension at 72 °C. The products of amplification were separated on a 1.6% (m/v) agarose gel, and bands were detected by ethidium bromide staining.
Amplified AFLP and ISSR fragments were scored as present (1) or absent (0) and recorded as binary matrices. XLSTAT Version 2016.01.26779 software (Addinsoft, Paris, France) was used to assess genetic variability between gametophytes and sporophytes before and after cryopreservation. The UPGMA dendrograms were constructed using Jaccard’s dissimilarity coefficient.
Results
Spore germination and gametophyte development
Freshly collected spores of A. cuneifolium germinated on average in 90% within 5 days from sowing in sterile, distilled water. The 1-year-long storage of spores at 5 °C ensured their viability in 80%, whereas storage in LN did not change it at all. Further 2-year-long storage in LN also allowed to maintain the initial viability of spores (Fig. 1).

The effect of the storage type and time on the survival of A. cuneifolium spores
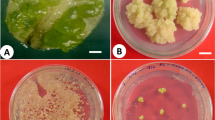
Spores disinfected with 5% Domestos started to germinate after 2 weeks from disinfection and sowing on GM (Fig. 2a). A few days later, young gametophytes developed (Fig. 2b-e). In the following weeks, the entire surface of the medium was covered with primary gametophytes (Fig. 2f), which proliferated easily and formed abundant clusters of secondary gametophytes within 6 months (Fig. 2g).

Germination of disinfected A. cuneifolium spores and development and multiplication of gametophytes: germination of a single spore after 2 weeks from sowing (a); subsequent cell divisions leading to development of a young gametophyte (b-e); culture of primary gametophytes on GM medium (f); intense proliferation of secondary gametophytes but without the appearance of sporophytes after 6 months of culture on GM medium (g)
Gametophyte survival after cryopreservation
The encapsulated, but non-frozen gametophytes of A. cuneifolium withstood PVS3 treatments, showing 100% survival regardless of treatment time (Fig. 3a). Cryopreservation significantly decreased the survival rate, which ranged from 64.5 to 93% depending on the time of previous incubation of gametophytes in PVS3. The highest survival of gametophytes was noted for 3-hour-long incubation (Fig. 3a). The recovery of gametophyte culture took about 5 weeks.

Cryopreservation of A. cuneifolium gametophytes: the effect of PVS3 treatment time on the survival of gametophytes in encapsulation-vitrification technique (a); the effect of the preculture on the survival of gametophytes in encapsulation-dehydration technique (b); limited portion of gametophyte surface that survived cryopreservation via encapsulation-dehydration without preculture (c); large portions of gametophyte surface which survived cryopreservation via encapsulation-dehydration with preculture (d); the recovery of gametophytes in post rewarming culture, 4 weeks after cryopreservation (e). Values followed by the same letters are not significantly different at p < 0.05 (HSD test)
The use of the encapsulation-dehydration technique for cryopreservation of gametophytes resulted in their 100% survival, either with or without preculture (Fig. 3b). However, without the preculture step, only limited gametophyte cells survived (Fig. 3c), whereas the implementation of the preculture reduced tissue necrosis (Fig. 3d) and shortened the time needed for regeneration of gametophytes (Fig. 3e) from 5 to 4 weeks.
Sporophyte formation via syngamy and SE
While growing on GM, gametophytes of A. cuneifolium proliferated but hardly ever produced sporophytes. The syngamy and development of the first sporophytes (Fig. 4a) were observed after 6 months of culture, and were more intense in senescent 12-month-old cultures maintained in the same medium without subculture. The reduction of macro and micronutrients in the medium combined with the complete withdrawal of NH4NO3 and vitamins allowed for a significantly faster sporophytogenesis. The appearance of sporophytes was evident after 5 weeks from the transfer of secondary gametophytes onto 1/8 MS medium (Fig. 4b). After cryopreservation, the reproduction of sporophytes proceeded similarly to that of control gametophyte cultures.

Development and multiplication of A. cuneifolium sporophytes via syngamy and somatic embryogenesis: very young sporophytes which developed via syngamy on gametophyte (a); numerous young sporophytes obtained via syngamy after 5 weeks of culture on GM medium devoid of ammonium ions and vitamins and with reduced macro- and micronutrients (b); etiolated stipe explant with first divisions (arrows) of epidermal cells perpendicular to the polar axis of the explant (c); successive cell divisions (arrows) leading to the formation of a linear pro-embryo from epidermal cells of stipe explants visible in ESEM (d) and epifluorescence microscopy (e); a multicellular pro-embryo which developed following several anticlinal, periclinal and inclined cell divisions of single epidermal cell of stipe explant (f); a stipe explant with several immature somatic embryos in the early embryonic leaf stage after 6 weeks of growth (g); somatic embryo in the early embryonic leaf stage with trichomes located on one side in epifluorescence (h) and ESEM microscopy (i); differentiation of the embryonic leaf in the late embryonic leaf stage of somatic embryo (j); secondary somatic embryo-derived juvenile sporophytes showing the first leaf and primordium of the second leaf (arrow) in ESEM (k) and stereo/dissecting microscopy (l)
In the SE, which was induced on stipe explants collected from young leaves of etiolated juvenile sporophytes, somatic embryos originated from epidermal cells, which started to divide within 2 weeks of the culture (Fig. 4c). After several first cell divisions perpendicular to the polar axis of the explant leading to the formation of the linear pro-embryo (Fig. 4d-e), cells begin to divide anticlinally, periclinally and inclinally. As a result, a multicellular structure developed (Fig. 4f-g). In the next step, trichomes appeared on one side of the immature, multicellular somatic embryo and the differentiation of the embryonic leaf began (Fig. 3h-i). Soon after the first frond was formed (Fig. 4j), the primordium of the second leaf became visible (Fig. 4k). Within 6 months, young sporophytes with two or three leaves developed. Secondary SE was easily induced on stipe explants collected from such sporophytes (Fig. 4l).
In control conditions on IM, the percentage of responding explants was over 96% (Fig. 5a), however, the number of somatic embryos regenerated per one explant was low and amounted on average 2.5 (Fig. 5b). The increase in pH to 6.8 did not significantly affect the efficiency of SE, whereas the higher pH (7.8) reduced both the percentage of responding explants and the number of somatic embryos per explant by half (Fig. 5a-b). Furthermore, the enhancement of the sucrose concentration in the medium up to 0.15 M did not influence the SE effectiveness (Fig. 5c-e). The increase in the number of somatic embryos regenerated from a single explant was observed only on the medium with sucrose concentration elevated to 0.3 M. The number of embryos per explant was almost 4-time greater than for other concentrations (Fig. 5d). However, a reduction in sucrose concentration from 0.3 to 0.06 M after 2 months of culture was essential to achieve further proper sporophyte development (Fig. 5f). Young sporophytes grew slowly and produced callus tissue at the base when left on the medium with 10% sucrose (Fig. 5g).

The influence of pH and sucrose concentration in the culture medium on SE efficiency: the effect of pH on the percentage of A. cuneifolium stipe explants exhibiting somatic embryogenesis (a) and on the average number of somatic embryos per explant (b); the effect of sucrose concentration on the percentage of A. cuneifolium stipe explants exhibiting somatic embryogenesis (c) and on the average number of somatic embryos per explant (d); young sporophytes obtained on stipe explants of A. cuneifolium after 4 months of culture on the medium containing 0.03 M sucrose (e), after 2 months of culture on 0.3 M sucrose followed by 2 months on 0.06 M sucrose (f), and after 4 months of culture on the medium containing 0.3 M sucrose (g). Values followed by the same letters are not significantly different at p < 0.05 (HSD test)
Nuclear DNA content
Flow cytometry revealed that non-cryopreserved (control) gametophytes contained 5.07 pg/1 C DNA (Table 3; Fig. 6a), while gametophytes after cryopreservation contained 5.09 pg/1 C DNA (Table 3; Fig. 6b). Both groups of sporophytes tested possessed approximately twofold the DNA content of the gametophytes, 10.04 pg and 10.02 pg for sporophytes obtained from control and cryopreserved gametophytes, respectively (Table 3; Fig. 6c-d). There was no significant difference in nuclear DNA content between control and cryopreserved plant material (Table 3).

Exemplary histograms of nuclear DNA content of nuclei isolated simultaneously from the leaves of Secale cereale ‘Dańkowskie’ (internal standard) and from A. cuneifolium control gametophytes GC (a), gametophytes after cryopreservation GLN (b), sporophytes obtained from control gametophytes SC (c), sporophytes obtained from gametophytes after cryopreservation SLN (d)
Molecular analysis of gametophytes and sporophytes after cryopreservation
For all DNA samples, all AFLP and ISSR primers gave scorable and reproducible band patterns (Online Resource 1a-b). Total 340 and 167 bands with an average of 56.7 and 13.9 per primer pair / single primer were obtained for AFLP (Table 1) and ISSR analysis (Table 2), respectively.
Both analyses revealed that there was little genetic variability among most of the DNA samples. AFLP proved to be a bit more powerful in generating polymorphic fragments, than the ISSR (12.3% of average polymorphism for AFLP vs. 10.2% for ISSR; Table 1). However, both marker systems showed similar levels of general sequence variation. Moreover, on either AFLP or ISSR dendrograms, six groups of individuals were recognized (Online Resource 2a-b).
Based on the results of AFLP analysis, the greatest dissimilarity was noted for one of the sporophytes obtained from cryopreserved gametophytes (SLN-7) and for one of the control gametophytes (GC-12). Their genetic dissimilarity from other individuals, expressed in the Jaccard coefficient, was 0.067 and 0.043, respectively. This dissimilarity resulted from the presence of 6 and 3 unique bands and the absence of 7 and 5 bands common for other individuals (Online Resource 2a). The remaining individuals did not have unique bands, but among them SC-8 and SLN-4 lacked 3 and 1 bands, respectively.
ISSR analysis revealed that the most distant sample was again GC-12. In its profiles, 5 unique bands were detected, while 3 common bands were not. The Jaccard dissimilarity coefficient, which reflected its genetic dissimilarity from other individuals, was 0.073 (Online Resource 2b).
The UPGMA dendrogram (Fig. 7) which resulted from joint AFLP and ISSR analyses, did not clearly separate neither control A. cuneifolium individuals from cryopreserved ones nor gametophytes from sporophytes. Most of the generated clusters included all types of individuals. Only the two most distant samples, GC-12 and SLN-7, were grouped separately.

A dendrogram of genetic dissimilarity between individual control gametophytes (GC), gametophytes after cryopreservation (GLN), sporophytes obtained from control gametophytes (SC), and sporophytes obtained from gametophytes after cryopreservation (SLN), generated by the UPGMA cluster analysis based on combined AFLP and ISSR molecular markers
Discussion
Viability of stored spores
The majority of fern species are propagated and cultured through spore germination and gametophyte production (Ballesteros 2011). Ferns from the genus Asplenium produce nongreen spores, which can be treated as orthodox seeds, i.e., they are supposed to be easily stored for a long time under dry conditions, either at ambient or low temperature. The longest storage time was reported for Asplenium serra Langsd. & Fisch. Its spores remained viable for 48 years (Lloyd and Klekowski 1970; Aragon and Pangua 2004) tested the viability of spores of A. septentrionale, A. adiantum-nigrum, and Asplenium ruta-muraria L. after 12 months of storage under dry and wet conditions at 20 °C, 5 °C and − 20 °C. They reported high percentages of germination for the first two species regardless of storage conditions, except for wet storage at -20 °C. However, there was a slight decrease in spore viability for most storage combinations compared to the viability of control spores. We also observed that after 1 year of storage at 5 °C, the viability of A. cuneifolium spores decreased from 90 to 80%.
Studies on spores stored at temperatures of a freezer (-10 to -30 °C) suggested that fern spores may have analogous physiologies to seeds classified in the intermediate storage category and their longevity is reduced due to crystallization of triacylglycerols (TAG; Ballesteros 2011). However, storage in LN can prevent damage because TAG crystallization is inhibited (Crane et al. 2006). In the case of A. cuneifolium, storage of spores in LN did not affect their initial viability for 3 years.
Culture and cryopreservation of gametophytes
Although storing spores is considered to be the easiest and cheapest method for ex situ fern conservation, the storage of gametophytes can also be useful if only a few spores are available (Ballesteros and Pence 2018). In such a case, in vitro spore sowing and multiplication of prothalli can provide a theoretically unlimited supply of uniform and highly regenerative gametophytic tissue (Pence 2018). Previous studies on other fern species indicated that only a small gametophyte fragment surviving cryopreservation is needed to regenerate the whole culture (Mikuła et al. 2011). As in earlier reports (Kromer et al. 2006; Marszał-Jagacka and Kromer 2011), primary and secondary gametophytes of A. cuneifolium grew well on ½MS medium with 0.06 M sucrose, which provided a lot of initial plant material for cryopreservation and studies on sporophyte formation.
Until now, cryopreservation strategies have been developed for gametophytes of only 25 fern species (Zimnoch-Guzowska et al. 2022), and the majority of them exploited encapsulation-dehydration techniques (Pence 2018; Mikuła et al. 2022a). A few attempts of vitrification without encapsulation failed due to insufficient protection of gametophyte structural integrity against harmful high concentrations of applied cryo-solutions (Mikuła et al. 2011; Makowski et al. 2016). Here, encapsulation-dehydration was compared with encapsulation-vitrification, which is less popular for fern gametophytes but also less time-consuming. The latter technique resulted in a survival rate significantly lower for Dicksonia fibrosa Col. and other tree ferns than encapsulation-dehydration (Mikuła et al. 2011), but comparable for Osmunda regalis L. after choosing an appropriate PVS solution and optimization of vitrification time (Mikuła et al. 2011,; Makowski et al. 2016). The survival of A. cuneifolium gametophytes after encapsulation-vitrification was only slightly worse than after encapsulation-dehydration, but, similarly to the above-mentioned species, the percentage of the tissue necrosis was much greater. As a result, the time of gametophyte recovery was extended by a week, thereby eliminating any potential savings in time.
Another possibility to shorten the duration of the cryopreservation procedure is the omission of the preculture step. The effect of preculture in the encapsulation-dehydration method of fern cryopreservation was studied for a dozen or so species (Mikuła et al. 2009, 2011; Pence 2014, 2015; Makowski et al. 2015). The preculture conducted on medium with ABA and elevated sucrose concentration (Mikuła et al. 2009, 2011), as well as the reduced size of alginate beads (Makowski et al. 2015; Mikuła et al. 2022a), significantly improved the survival of gametophytes, especially those of sensitive species with low tolerance to LN exposure. On the other hand, gametophytes of A. scolopendrium var. americanum showed 75–100% survival, whether or not they were precultured on ABA-containing medium (Pence 2015). The preculture step on ABA-supplemented medium was not crucial for survival of gametophytes of A. scolopendrium var. scolopendrium either, but it significantly lowered the level of tissue necrosis and shortened the time required for complete recovery of gametophytes in post-thawing culture (Mikuła et al. 2011). Similarly, preculture was found to be non-obligatory to ensure the survival of A. cuneifolium gametophytes; however, its implementation reduced both the tissue necrosis and the time needed for regeneration of gametophytes by about a week.
Development and multiplication of sporophytes
The formation of Asplenium sporophytes via syngamy in tissue culture was studied by Kromer et al. (2006), Somer et al. (2010), and Marszał-Jagacka and Kromer (2011). According to their reports, development of a few single sporophytes has taken at least 2 months since transferring gametophytes onto fresh ½MS. In all studies, an imbalance in the number of antheridia and archegonia that developed on gametophytes was observed and regarded as a probable limiting factor in the fertilization and development of sporophytes. Research on other fern species like Pteris ensiformis Burm. f., Adiantum capillus-veneris, O. regalis or Alsophila costularis showed that a significant increase in the sporophyte formation could take place when gametophytes were cultured in media with a lowered concentration of mineral salts and vitamins or at least in the absence or reduced concentration of ammonium nitrate (Fernández et al. 1999; Kuriyama et al. 2004; Makowski et al. 2016; Pu et al. 2023). In our study, the use of a medium without NH4NO3 and vitamins and with macro- and micronutrients reduced to 1/8 MS allowed to shorten the time of sporophyte production from 6 months to 5 weeks.
Multiplication of sporophytes of epilithic Asplenium species has been performed mainly in three ways. First of them included the culture of sporophytes in MS medium supplemented with 4.4 µM BAP, and then the transfer of swelling rhizomes to medium devoid of PGRs. Such rhizomes formed numerous fronds (Somer et al. 2010). The second approach involved homogenization of sporophytes and subsequent culture in a liquid MS medium with the addition of BAP. This encouraged the formation of well-organized structures, either aposporous gametophytes or sporophytes of A. adiantum-nigrum (Somer et al. 2010). The third way was to culture single, separated rhizomes on medium supplemented with low concentrations of NAA and kinetin, which stimulated the development of new fronds (Marszał-Jagacka and Kromer 2011). However, regeneration of sporophytes was also observed on fronds inoculated horizontally on ½MS medium supplemented with 0.58 µM IAA and 4.65 or 9.3 µM kinetin. Interestingly, when frond stipes were wounded, the regeneration process occurred on a medium devoid of PGRs (Marszał-Jagacka and Kromer 2011). This suggests that the stipe explants possess morphogenetic abilities, which can be activated after applying wounding stress. In previous studies, we achieved highly efficient somatic embryogenesis on stipe explants excised from etiolated fronds of the tree fern C. delgadii (Mikuła et al. 2015a, b) without applying any PGRs. A similar process was also initiated here, on stipe explants of A. cuneifolium. Moreover, the same developmental stages as in the case of C. delgadii, i.e., linear pro-embryo stage, as well as early and late embryonic leaf stages (Mikuła et al. 2015b), were recognized. Although stipe explants of A. cuneifolium regenerated fewer somatic embryos than those of C. delgadii, a higher percentage of responding explants (over 96%) was observed. Also, in contrast to C. delgadii, for which the efficiency of SE decreased with the increase in sucrose concentration in the medium up to 0.15 M (Mikuła et al. 2015a) and the process was completely inhibited after short treatments of stipe explants with highly concentrated (over 0.4 M) sucrose solutions (Grzyb and Mikuła 2019), A. cuneifolium stipes reacted indifferently to the concentration of sucrose in the culture medium reaching 0.15 M, whereas the addition of 0.3 M sucrose resulted in the number of embryos per explant almost 4-time greater than for other concentrations tested.
Genetic stability of cryopreserved gametophytes
For numerous plant species, cryopreservation technology is inseparable from in vitro culture methods used for the establishment and maintenance of initial plant material, its preculture, and the process of recovery and regrowth of tissues in post-thawing culture (Wang et al. 2021). However, plant tissue culture is known to engender frequent unwanted genetic and epigenetic variation, such as chromosomal and DNA sequence variation, methylation changes or activation of transposons (Neelakandan and Wang 2012; Fossi et al. 2019). Thus, a thorough analysis of the genetic stability of regenerated plants should be an indispensable part of studies aimed at developing regeneration and cryopreservation systems for species of interest. However, analysis of somaclonal variation has been reported very occasionally for ferns. To the best of our knowledge, only Marsilea quadrifolia L. propagated in vitro from nodal explants has been examined using isoenzyme (Banciu et al. 2009) and RAPD markers (Rolli et al. 2015). The results of isozyme analyses showed more intense catalytic activity in regenerants than in native plants (Banciu et al. 2009), whereas no genomic alterations were evidenced in any of the micropropagated plants by RAPD markers (Rolli et al. 2015). Among fern species subjected to cryopreservation, only Cyathea australis (R. BR.) Domin has been analyzed, and it was done only with the use of flow cytometry (Mikuła et al. 2009). In this study, significant differences between non- and cryopreserved plant material were detected.
The nuclear DNA content obtained for A. cuneifolium was in accordance with previous studies by Clark et al. (2016) and Siljak-Yakovlev et al. (2010), who reported from 5.07 to 5.11 pg/1 C for gametophytes of this species growing in various natural populations. The lack of statistically significant differences between the DNA content of control gametophytes and gametophytes after cryopreservation as well as between the DNA content of sporophytes obtained from control and cryopreserved gametophytes suggest that the applied protocol of cryopreservation did not introduce essential quantitative changes at the chromosome level. Moreover, the DNA content recorded for both tested groups of sporophytes confirms their syngamic origin.
In order to exploit as many sources of polymorphism as possible and provide more reliable results of genetic integrity assessments, it is recommended to use more than one molecular technique. This is because various DNA markers target different regions of the genome (Velasco-Ramírez et al. 2014; Costa et al. 2016; Wang et al. 2021). The most popular markers for rapid detection of polymorphism and generation of reproducible fragments without prior sequence information are AFLP (Agarwal et al. 2008; Meudt and Clarke 2007) and ISSR (Pradeep Reddy et al. 2002).
The lack of appropriate literature data concerning molecular analysis of gametophytes prompted us to hypothesize that genetic variation of plant material taken for cryopreservation, i.e., gametophytes obtained from spores derived from one leaf, should be minimal. However, one of the gametophytes (GC-12) turned out to be significantly different from the rest, possessing 8 unique and lacking 8 common bands. This variation in DNA sequence was probably not induced by in vitro culture conditions, but pre-existed in plant material, since the protocol for spore sowing and gametophyte culture was simple and quick without employing callus phase and PGR-supplemented media, which are well-known factors favoring somaclonal variation (Bairu et al. 2011).
The genetic variation detected in plant material obtained after cryopreservation with the use of AFLP and ISSR markers mainly reflected variation that was already present in cryopreserved material. Most polymorphic bands were also found in electrophoretic patterns of control gametophytes and sporophytes. However, although the protocol applied here minimized the likelihood of genetic changes since it did not require the use of cryoprotectants and no callus was formed in that time (Wang et al. 2014), it cannot be excluded that sequence variation observed for sporophytes SLN-4 (lack of 1 common band) and SLN-7 (presence of 6 and lack of 7 common bands) resulted from the cryopreservation procedure.
Conclusion
The presented studies were aimed at evaluating the potential of in vitro propagation and cryopreservation to assist long-term conservation of the endangered serpentine fern A. cuneifolium. We showed that spores can be safely stored in LN without losing their viability and, after disinfection, they can be used to initiate an in vitro culture of gametophytes. We also demonstrated that cryopreservation of gametophytes using the encapsulation-dehydration method guarantees their 100% survival and the implementation of the preculture step on a medium supplemented with 10 µM ABA and 0.25 M sucrose allows shortening the time of their recovery in post-rewarming culture. The development of sporophytes via syngamy can be accelerated by the reduction of macro- and micronutrients and the withdrawal of NH4NO3 and vitamins in the medium, and their multiplication is possible via somatic embryogenesis without using PGRs. Despite some minimal changes in the DNA sequence, the applied cryopreservation protocol does not disturb the nuclear DNA content of gametophytes and sporophytes.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Agarwal M, Shrivastava N, Padh H (2008) Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep 27:617–631. https://doi.org/10.1007/s00299-008-0507-z
Aldea F, Banciu C, Brezeanu A, Helepciuc FE, Soare L-C (2016) In vitro micropropagation of fern species (Pteridophyta) of biotechnological interest, for ex situ conservation. Oltenia Stud şi comunicări Ştiinţele Nat 32:27–35
Aragon CF, Pangua E (2004) Spore viability under different storage conditions in four rupicolous Asplenium L. taxa. Am Fern J 94:28–38. https://doi.org/10.1640/0002-8444(2004)094[0028:SVUDSC]2.0.CO;2
Bairu MW, Aremu AO, van Staden J (2011) Somaclonal variation in plants: causes and detection methods. Plant Growth Regul 63:147–173. https://doi.org/10.1007/s10725-010-9554-x
Ballesteros D (2011) Conservation of fern spores. In: Fernandez H, Kumar A, Revilla MA (eds) Working with ferns: issues and applications. Springer Science + Business Media, New York, pp 165–172. https://doi.org/10.1007/978-1-4419-7162-3_12
Ballesteros D, Pence VC (2018) Fern conservation: spore, gametophyte, and sporophyte ex situ storage, in vitro culture, and cryopreservation. In: Fernández H (ed) Current advances in Fern Research. Springer International Publishing AG, pp 227–249. https://doi.org/10.1007/978-3-319-75103-0_11
Banciu C, Carasan ME, Brezeanu A (2009) In vitro propagation of the endangered species Marsilea quadrifolia L. – Morphological and biochemical analysis of the regenerates. Rom Biotechnol Lett 14:4139–4145
Barnicoat H, Cripps R, Kendon J, Sarasan V (2011) Conservation in vitro of rare and threatened ferns – case studies of biodiversity hotspot and island species. In Vitro Cell Dev Biol—Plant 47:37–45. https://doi.org/10.1007/s11627-010-9303-x
Bucharová A, Münzbergová Z, Tájek P (2010) Population biology of two rare fern species: long life and long-lasting stability. Am J Bot 97:1260–1271. https://doi.org/10.3732/ajb.0900351
Bunn E, Turner S, Panaia M, Dixon KW (2007) The contribution of in vitro technology and cryogenic storage to conservation of indigenous plants. Aust J Bot 55:345–355. https://doi.org/10.1071/BT06065
Clark J, Hidalgo O, Pellicer J, Liu H, Marquardt J, Robert Y, Christenhusz M, Zhang S, Gibby M, Leitch IJ, Schneider H (2016) Genome evolution of ferns: evidence for relative stasis of genome size across the fern phylogeny. New Phytol 210:1072–1082. https://doi.org/10.1111/nph.13833
Coelho N, Gonçalves S, Romano A (2020) Endemic plant species conservation: biotechnological approaches. Plants 9:345. https://doi.org/10.3390/plants9030345
Costa R, Pereira G, Garrido I, Tavares-De-Sousa MM, Espinosa F (2016) Comparison of RAPD, ISSR, and AFLP molecular markers to reveal and classify Orchardgrass (Dactylis glomerata L.) germplasm variations. PLoS ONE 11:1–15. https://doi.org/10.1371/journal.pone.0152972
Crane J, Kovach D, Gardner C, Walters C (2006) Triacylglycerol phase and “intermediate” seed storage physiology: a study of Cuphea carthagenensis. Planta 223:1081–1089. https://doi.org/10.1007/s00425-005-0157-0
Doležel J, Greilhuber J, Lucretti S, Meister A, Lysák MA, Nardi L, Obermayer R (1998) Plant genome size estimation by flow cytometry: inter-laboratory comparison. Ann Bot 82:17–26. https://doi.org/10.1006/anbo.1998.0730
Fernández H, Bertrand A, Sánchez-Tamés R (1993) In vitro regeneration of Asplenium nidus L. from gametophytic and sporophytic tissue. Sci Hortic (Amsterdam) 56:71–77. https://doi.org/10.1016/0304-4238(93)90103-W
Fernández H, Bertrand A, Sánchez-Tamés R (1997) Plantlet regeneration in Asplenium nidus L. and Pteris ensiformis L. by homogenization of BA treated rhizomes. Sci Hortic (Amsterdam) 68:243–247. https://doi.org/10.1016/S0304-4238(96)00986-7
Fernández H, Bertrand AM, Sánchez-Tamés R (1999) Biological and nutritional aspects involved in fern multiplication. Plant Cell Tissue Organ Cult 56:211–214. https://doi.org/10.1023/A:1006277229136
Fossi M, Amundson K, Kuppu S, Britt A, Comai L (2019) Regeneration of Solanum tuberosum plants from protoplasts induces widespread genome instability. Plant Physiol 180:78–86. https://doi.org/10.1104/pp.18.00906
Grzyb M, Mikuła A (2019) Explant type and stress treatment determine the uni- and multicellular origin of somatic embryos in the tree fern Cyathea delgadii Sternb. Plant Cell Tissue Organ Cult 136:221–230. https://doi.org/10.1007/s11240-018-1507-5
Higuchi H, Amaki W (1989) Effects of 6-benzylaminopurine on the organogenesis of Asplenium nidus L. through in vitro propagation. Sci Hortic (Amsterdam) 37:351–359. https://doi.org/10.1016/0304-4238(89)90146-5
Kaźmierczakowa R, Bloch-Orlowska J, Celka Z, Cwener A, Dajdok Z, Michalska-Hejduk D, Pawlikowski P, Szczęśniak E, Ziarnek K (2016) Polish red list of pteridophytes and flowering plants. Institute of Nature Conservation PAS, Kraków
Khan S, Raziq M, Kayani HA (2008) In vitro propagation of bird’s nest fern (Asplenium nidus) from spores. Pakistan J Bot 40:91–97
Kromer K, Marszał-Jagacka J, Kempińska K, Nowak T, Żołnierz L, Poturała D, Świerkosz K (2006) In vitro propagation and ex situ preservation of endangered ferns from Lower Silesia. Bot Guid 29:143–155
Kuriyama A, Kobayashi T, Hayashi S, Maeda M (2004) Medium composition for the production of sporophytes of the fern Adiantum capillus-veneris. J Japanese Soc Hortic Sci 73:580–582. https://doi.org/10.2503/jjshs.73.580
Lin Y, Viane R (2014) Aspleniaceae. In: Wu ZY, Raven PH, Hong DY (eds) Flora of China, Vols. 2–3, Lycopodiaceae through Polypodiaceae. Science Press; St. Louis: Missouri Botanical Garden Press., Beijing, St. Louis, Beijing, pp 267–316
Lloyd RM, Klekowski EJ (1970) Spore germination and viability in Pteridophyta: evolutionary significance of chlorophyllous spores. Biotropica 2:129–137. https://doi.org/10.2307/2989770
Makowski D, Rybczyński JJ, Mikuła A (2015) A simple way to overcome the recalcitrance of the water fern Ceratopteris thalictroides (L.) Brongn. To cryopreservation. Acta Soc Bot Pol 84:385–388. https://doi.org/10.5586/asbp.2015.032
Makowski D, Tomiczak K, Rybczyński JJ, Mikuła A (2016) Integration of tissue culture and cryopreservation methods for propagation and conservation of the fern Osmunda regalis L. Acta Physiol Plant 38:19. https://doi.org/10.1007/s11738-015-2037-y
Marszał-Jagacka J, Kromer K (2011) In vitro propagation of rare and endangered serpentine fern species. In: Kumar A, Fernández H, Revilla M (eds) Working with ferns: issues and applications. Springer Science + Business Media, pp 149–164. https://doi.org/10.1007/978-1-4419-7162-3_11
Marszał-Jagacka J, Kromer K, Świerkosz K (2005) Ex situ protection of endangered Asplenium ferns species using in vitro culture. Biul Ogrodów Bot Muzeów i Zbior 14:35–42
Mehltreter K (2010) Fern conservation. In: Mehltreter K, Walker LR, Sharpe JM (eds) Fern Ecology. Cambridge University Press, Cambridge, pp 323–359. https://doi.org/10.1017/CBO9780511844898.010
Menéndez V, Abul Y, Bohanec B, Lafont F, Fernández H (2011) The effect of exogenous and endogenous phytohormones on the in vitro development of gametophyte and sporophyte in Asplenium nidus L. Acta Physiol Plant 33:2493–2500. https://doi.org/10.1007/s11738-011-0794-9
Meudt HM, Clarke AC (2007) Almost Forgotten or Latest Practice? AFLP applications, analyses and advances. Trends Plant Sci 12:106–117. https://doi.org/10.1016/j.tplants.2007.02.001
Mikuła A, Jata K, Rybczyński JJ (2009) Cryopreservation strategies for Cyathea australis (R. Br.) Domin. Cryo-Letters 30:429–439
Mikuła A, Makowski D, Walters C, Rybczyński JJ (2011) Exploration of cryo-methods to preserve tree and herbaceous fern gametophytes. In: Fernández H, Kumar A, Revilla A (eds) Working with ferns: issues and applications. Springer, New York, pp 173–192. https://doi.org/10.1007/978-1-4419-7162-3_13
Mikuła A, Pożoga M, Grzyb M, Rybczyński JJ (2015a) An unique system of somatic embryogenesis in the tree fern Cyathea delgadii Sternb.: the importance of explant type, and physical and chemical factors. Plant Cell Tissue Organ Cult 123:467–478. https://doi.org/10.1007/s11240-015-0850-z
Mikuła A, Pożoga M, Tomiczak K, Rybczyński JJ (2015b) Somatic embryogenesis in ferns: a new experimental system. Plant Cell Rep 34:783–794. https://doi.org/10.1007/s00299-015-1741-9
Mikuła A, Tomiczak K, Makowski D, Niedzielski M, Rybczyński JJ (2015c) The effect of moisture content and temperature on spore aging in Osmunda regalis. Acta Physiol Plant 37:229. https://doi.org/10.1007/s11738-015-1985-6
Mikuła A, Chmielarz P, Hazubska-Przybył T, Kulus D, Maślanka M, Pawłowska B, Zimnoch-Guzowska E (2022a) Cryopreservation of plant tissues in Poland: Research contributions, current status, and applications. Acta Soc Bot Pol 91:9132. https://doi.org/10.5586/asbp.9132
Mikuła A, Gaj M, Grzyb M, Hazubska-Przybył T, Kępczyńska E, Kępczyński J, Rybczyński J, Tomiczak K, Wójcik AM (2022b) Polish contribution to global research on somatic embryogenesis. Acta Soc Bot Pol 91:9115. https://doi.org/10.5586/asbp.9115
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Neelakandan AK, Wang K (2012) Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications. Plant Cell Rep 31:597–620. https://doi.org/10.1007/s00299-011-1202-z
Nishizawa S, Sakai A, Amano Y, Matsuzawa T (1993) Cryopreservation of asparagus (Asparagus officinalis L.) embryogenic suspension cells and subsequent plant regeneration by vitrification. Plant Sci 91:67–73. https://doi.org/10.1016/0168-9452(93)90189-7
Pence VC (2014) In vitro propagation and cryopreservation of the endangered filmy fern, Trichomanes punctatum subsp. floridanum (Hymenophyllaceae). Fern Gaz 19:307–317
Pence VC (2015) Propagation and cryopreservation of Asplenium scolopendrium var. americanum, the American hart’s-tongue fern. Am Fern J 105:211–225. https://doi.org/10.1640/0002-8444-105.3.211
Pence VC (2018) Growth of fern gametophytes after 20 years of storage in liquid nitrogen. Fern Gaz 20:337–346
Pradeep Reddy M, Sarla N, Siddiq EA (2002) Inter simple sequence repeat (ISSR) polymorphism and its application in plant breeding. Euphytica 128:9–17. https://doi.org/10.1023/A:1020691618797
Pu Y, Song Q, Wang G, Wu L, Yang C, Yu R (2023) In vitro propagation and long-term observation of acclimated plants in endangered tree fern Alsophila costularis. Plant Cell, Tissue Organ Cult. 152:275–285. https://doi.org/10.1007/s11240-022-02402-5
Rolli E, Brunoni F, Marieschi M, Torelli A, Ricci A (2015) In vitro micropropagation of the aquatic fern Marsilea quadrifolia L. and genetic stability assessment by RAPD markers. Plant Biosyst 149:7–14. https://doi.org/10.1080/11263504.2013.806366
Siljak-Yakovlev S, Pustahija F, Šolić EM, Bogunić F, Muratović E, Bašić N, Catrice O, Brown SC (2010) Towards a genome size and chromosome number database of balkan flora: C-values in 343 taxa with novel values for 242. Adv Sci Lett 3:190–213. https://doi.org/10.1166/asl.2010.1115
Smith AR, Pryer KM, Schuettpelz E, Korall P, Schneider H, Wolf PG (2006) A classification for extant ferns. Taxon 55:705–731. https://doi.org/10.2307/25065646
Somer M, Arbesú R, Menéndez V, Revilla MA, Fernández H (2010) Sporophyte induction studies in ferns in vitro. Euphytica 171:203–210. https://doi.org/10.1007/s10681-009-0018-1
Szczęśniak E (2006) Asplenium serpentine ferns in Poland – threats and conservation imperatives. Bot Guid 29:89–97
Tomiczak K, Sliwinska E, Rybczyński JJ (2017) Protoplast fusion in the genus Gentiana: genomic composition and genetic stability of somatic hybrids between Gentiana kurroo Royle and G. cruciata L. Plant Cell Tiss Organ Cult 131:1–14. https://doi.org/10.1007/s11240-017-1256-x
Tomiczak K, Grzyb M, Rybczyński JJ, Mikuła A (2018) Somatic embryogenesis and somatic embryo cryopreservation of the tree-fern Cyathea delgadii Sternb. In: Jain SM, Gupta P (eds) Step Wise Protocols for somatic embryogenesis of important Woody plants. Springer International Publishing, pp 291–303. https://doi.org/10.1007/978-3-319-79087-9_23
Velasco-Ramírez AP, Torres-Morán MI, Molina-Moret S, de Jesús Sánchez-González J, Santacruz-Ruvalcaba F (2014) Efficiency of RAPD, ISSR, AFLP and ISTR markers for the detection of polymorphisms and genetic relationships in camote de cerro (Dioscorea spp). Electron J Biotechnol 17:65–71. https://doi.org/10.1016/j.ejbt.2014.01.002
Wang B, Wang RR, Cui ZH, Bi WL, Li JW, Li BQ, Ozudogru EA, Volk GM, Wang QC (2014) Potential applications of cryogenic technologies to plant genetic improvement and pathogen eradication. Biotechnol Adv 32:583–595. https://doi.org/10.1016/j.biotechadv.2014.03.003
Wang MR, Bi W, Shukla MR, Ren L, Hamborg Z, Blystad DR, Saxena PK, Wang QC (2021) Epigenetic and genetic integrity, metabolic stability, and field performance of cryopreserved plants. Plants 10:1–19. https://doi.org/10.3390/plants10091889
Xu KW, Zhang L, Rothfels CJ, Smith AR, Viane R, Lorence D, Wood KR, Chen CW, Knapp R, Zhou L, Lu NT, Zhou XM, Wei HJ, Fan Q, Chen SF, Cicuzza D, Gao XF, Liao WB, Zhang LB (2020) A global plastid phylogeny of the fern genus Asplenium (Aspleniaceae). Cladistics 36:22–71. https://doi.org/10.1111/cla.12384
Young BA, Sherwood RT, Bashaw EC (1979) Cleared-pistil and thick-sectioning techniques for detecting aposporous apomixis in grasses. Can J Bot 57:1668–1672. https://doi.org/10.1139/b79-204
Zimnoch-Guzowska E, Chmielarz P, Wawrzyniak MK, Plitta-Michalak BP, Michalak M, Pałucka M, Wasileńczyk U, Kosek P, Kulus D, Rucińska A, Mikuła A (2022) Polish cryobanks: Research and conservation of plant genetic resources. Acta Soc Bot Pol 91:9121. https://doi.org/10.5586/asbp.9121
Acknowledgements
We thank Małgorzata Grzyb for help with the plant material clearing in methyl salicylate and image capturing. This work was partially supported by the National Science Centre (NCN) in Poland, No. N N304 175540.
Funding
This work was partially supported by the National Science Centre (NCN) in Poland, No. N N304 175540.
Author information
Authors and Affiliations
Contributions
Karolina Tomiczak designed and performed the experiments concerning somatic embryogenesis and genetic stability after cryopreservation, analyzed the data and wrote the paper; Damian Makowski performed the experiments concerning in vitro culture and cryopreservation; Elwira Sliwinska performed flow cytometry and analyzed the data; Anna Mikuła designed the experiments concerning in vitro culture and cryopreservation and supervised the whole project. All authors were involved in editing the manuscript and approved its final version.
Corresponding author
Ethics declarations
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
Conflict of interest
The authors declare no conflict of interest exists.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by Ranjith Pathirana.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tomiczak, K., Makowski, D., Sliwinska, E. et al. The development of an in vitro propagation and conservation system for the endangered serpentine fern Asplenium cuneifolium Viv.. Plant Cell Tiss Organ Cult 154, 161–175 (2023). https://doi.org/10.1007/s11240-023-02524-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-023-02524-4