
A Novel Phenotype of Junctional Epidermolysis Bullosa with Transient Skin Fragility and Predominant Ocular Involvement Responsive to Human Amniotic Membrane Eyedrops

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
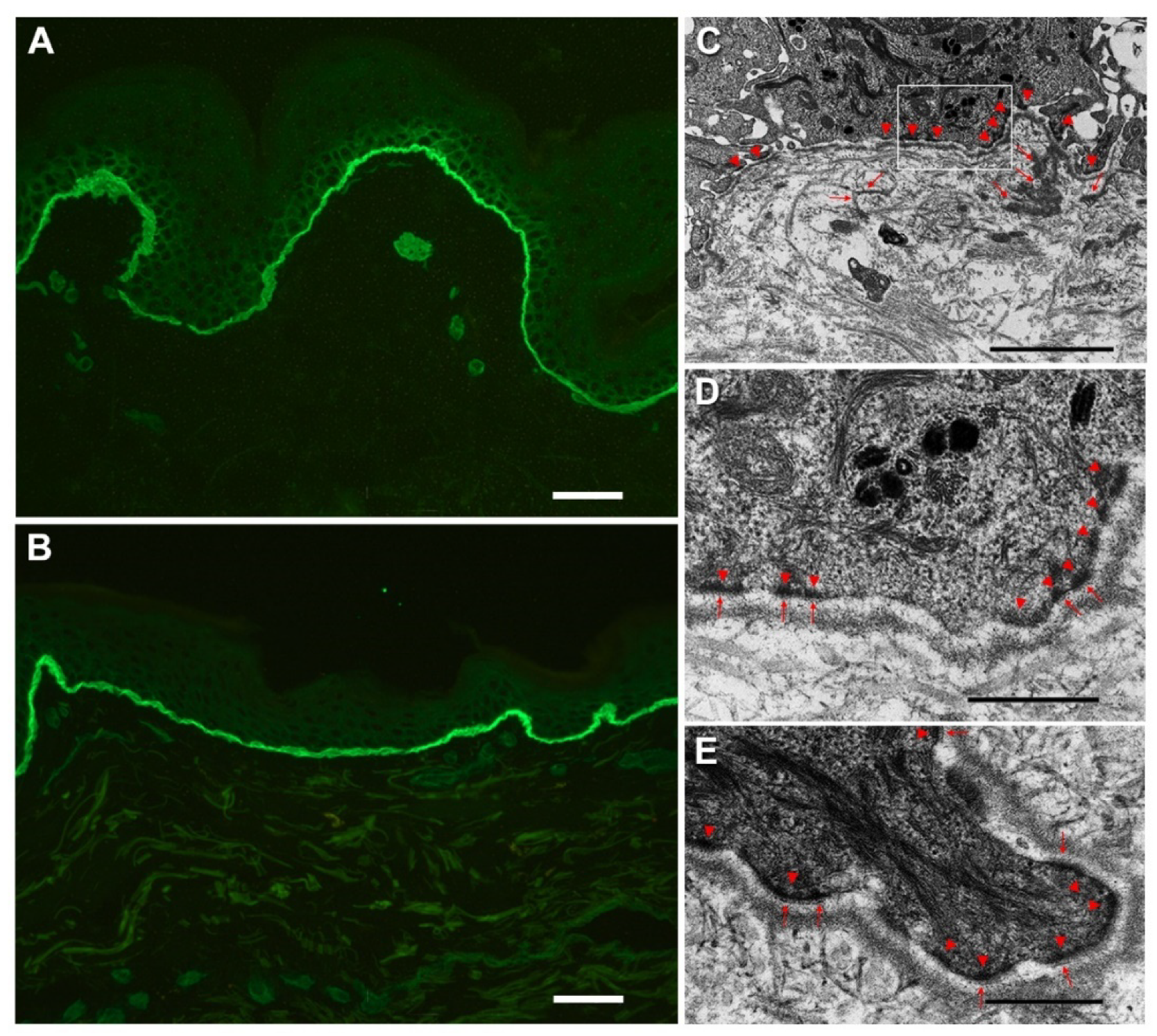{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Immunofluorescence and Ultrastructural Analyses and Cell Cultures
2.2. AM Eyedrop Preparation
2.3. DNA and RNA Studies
2.4. Immunoprecipitation and Immunoblotting Assays
2.5. Trypsin-Detachment Assays
3. Results
3.1. A Novel JEB Phenotype with Transient Skin Fragility and Severe Ocular Involvement
3.2. Compound Heterozygosity for Readthrough and Splice Site LAMB3 Variants Underlies a Novel JEB Phenotype
3.3. c.3052-5C>G Results in Residual Secretion of Wild-Type LM332
3.4. An AM Eyedrop Preparation Induces Long-Lasting Remission of Corneal Abrasions and Increases Patient Keratinocyte Adhesion.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Has, C.; Nyström, A.; Saeidian, A.H.; Bruckner-Tuderman, L.; Uitto, J. Epidermolysis bullosa: Molecular pathology of connective tissue components in the cutaneous basement membrane zone. Matrix Biol. 2018, 71–72, 313–329. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [Green Version]
- Rousselle, P.; Beck, K. Laminin 332 processing impacts cellular behavior. Cell Adhes. Migr. 2013, 7, 122–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, C.; Wang, C.K.; Nelson, C.F.; Bauer, E.A.; Hoeffler, W.K. The assembly of laminin-5 subunits. J. Biol. Chem. 1995, 270, 23496–23503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiritsi, D.; Has, C.; Bruckner-Tuderman, L. Laminin 332 in junctional epidermolysis bullosa. Cell Adhes. Migr. 2013, 7, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, A.; Chao, S.C.; Pulkkinen, L.; Murrell, D.; Bruckner-Tuderman, L.; Pfendner, E.; Uitto, J. Laminin 5 mutations in junctional epidermolysis bullosa: Molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum. Genet. 2002, 110, 41–51. [Google Scholar] [CrossRef]
- Posteraro, P.; De Luca, N.; Meneguzzi, G.; El Hachem, M.; Angelo, C.; Gobello, T.; Tadini, G.; Zambruno, G.; Castiglia, D. Laminin-5 mutational analysis in an Italian cohort of patients with junctional epidermolysis bullosa. J. Investig. Dermatol. 2004, 123, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiritsi, D.; Huilaja, L.; Franzke, C.W.; Kokkonen, N.; Pazzagli, C.; Schwieger-Briel, A.; Larmas, M.; Bruckner-Tuderman, L.; Has, C.; Tasanen, K. Junctional epidermolysis bullosa with LAMB3 splice-site mutations. Acta Derm. Venereol. 2015, 95, 849–851. [Google Scholar] [CrossRef]
- Fortugno, P.; Condorelli, A.G.; Dellambra, E.; Guerra, L.; Cianfarani, F.; Tinaburri, L.; Proto, V.; De Luca, N.; Passarelli, F.; Ricci, F.; et al. Multiple Skin Squamous Cell Carcinomas in Junctional Epidermolysis Bullosa Due to Altered Laminin-332 Function. Int. J. Mol. Sci. 2020, 21, 1426. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Stein, A.; Cash, S.; Deleoz, J.; Devries, D.T.; Suchindran, C. Eye involvement in inherited epidermolysis bullosa: Experience of the National Epidermolysis Bullosa Registry. Am. J. Ophthalmol. 2004, 138, 254–262. [Google Scholar] [CrossRef]
- Lin, A.N.; Murphy, F.; Brodie, S.E.; Carter, D.M. Review of ophthalmic findings in 204 patients with epidermolysis bullosa. Am. J. Ophthalmol. 1994, 118, 384–390. [Google Scholar] [CrossRef]
- Tong, L.; Hodgkins, P.R.; Denyer, J.; Brosnahan, D.; Harper, J.; Russell-Eggitt, I.; Taylor, D.S.; Atherton, D. The eye in epidermolysis bullosa. Br. J. Ophthalmol. 1999, 83, 323–326. [Google Scholar] [CrossRef]
- Jonsson, F.; Byström, B.; Davidson, A.E.; Backman, L.J.; Kellgren, T.G.; Tuft, S.J.; Koskela, T.; Rydén, P.; Sandgren, O.; Danielson, P.; et al. Mutations in collagen, type XVII, α 1 (COL17A1) cause epithelial recurrent erosion dystrophy (ERED). Hum. Mutat. 2015, 36, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Oliver, V.F.; van Bysterveldt, K.A.; Cadzow, M.; Steger, B.; Romano, V.; Markie, D.; Hewitt, A.W.; Mackey, D.A.; Willoughby, C.E.; Sherwin, T.; et al. A COL17A1 Splice-Altering Mutation Is Prevalent in Inherited Recurrent Corneal Erosions. Ophthalmology 2016, 123, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Leal-Marin, S.; Kern, T.; Hofmann, N.; Pogozhykh, O.; Framme, C.; Börgel, M.; Figueiredo, C.; Glasmacher, B.; Gryshkov, O. Human Amniotic Membrane: A review on tissue engineering, application, and storage. J. Biomed. Mater. Res. B Appl. Biomater. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rama, P.; Giannini, R.; Bruni, A.; Gatto, C.; Tiso, R.; Ponzin, D. Further evaluation of amniotic membrane banking for transplantation in ocular surface diseases. Cell Tissue Bank. 2001, 2, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Paolin, A.; Cogliati, E.; Trojan, D.; Griffoni, C.; Grassetto, A.; Elbadawy, H.M.; Ponzin, D. Amniotic membranes in ophthalmology: Long term data on transplantation outcomes. Cell Tissue Bank. 2016, 17, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Terracina, M.; Posteraro, P.; Schubert, M.; Sonego, G.; Atzori, F.; Zambruno, G.; Bruckner-Tuderman, L.; Castiglia, D. Compound heterozygosity for a recessive glycine substitution and a splice site mutation in the COL7A1 gene causes an unusually mild form of localized recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 1998, 111, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Zambruno, G.; Marchisio, P.C.; Marconi, A.; Vaschieri, C.; Melchiori, A.; Giannetti, A.; De Luca, M. Transforming growth factor-β 1 modulates β 1 and β 5 integrin receptors and induces the de novo expression of the α v β 6 heterodimer in normal human keratinocytes: Implications for wound healing. J. Cell Biol. 1995, 129, 853–865. [Google Scholar] [CrossRef]
- Posteraro, P.; Sorvillo, S.; Gagnoux-Palacios, L.; Angelo, C.; Paradisi, M.; Meneguzzi, G.; Castiglia, D.; Zambruno, G. Compound heterozygosity for an out-of-frame deletion and a splice site mutation in the LAMB3 gene causes nonlethal junctional epidermolysis bullosa. Biochem. Biophys. Res. Commun. 1998, 243, 758–764. [Google Scholar] [CrossRef]
- Condorelli, A.G.; Fortugno, P.; Cianfarani, F.; Proto, V.; Di Zenzo, G.; Didona, B.; Zambruno, G.; Castiglia, D. Lack of K140 immunoreactivity in junctional epidermolysis bullosa skin and keratinocytes associates with misfolded laminin epidermal growth factor-like motif 2 of the β3 short arm. Br. J. Dermatol. 2018, 178, 1416–1422. [Google Scholar] [CrossRef]
- Löffek, S.; Hurskainen, T.; Jackow, J.; Sigloch, F.C.; Schilling, O.; Tasanen, K.; Bruckner-Tuderman, L.; Franzke, C.W. Transmembrane collagen XVII modulates integrin dependent keratinocyte migration via PI3K/Rac1 signaling. PLoS ONE 2014, 9, e87263. [Google Scholar] [CrossRef] [PubMed]
- Torricelli, A.A.; Singh, V.; Santhiago, M.R.; Wilson, S.E. The corneal epithelial basement membrane: Structure, function, and disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6390–6400. [Google Scholar] [CrossRef]
- Nakao, M.; Kim, K.; Nagase, K.; Grainger, D.W.; Kanazawa, H.; Okano, T. Phenotypic traits of mesenchymal stem cell sheets fabricated by temperature-responsive cell culture plate: Structural characteristics of MSC sheets. Stem Cell Res. Ther. 2019, 10, 353. [Google Scholar] [CrossRef]
- Takashima, S.; Fujita, Y.; Shinkuma, S.; Shimizu, S.; Hasegawa, T.; Amizuka, N.; Shimizu, H.; Natsuga, K. Calcinosis cutis in self-healing dominant dystrophic epidermolysis bullosa. J. Dermatol. 2020, 47, e457–e58. [Google Scholar] [CrossRef]
- Utani, A.; Nomizu, M.; Timpl, R.; Roller, P.P.; Yamada, Y. Laminin chain assembly. Specific sequences at the C terminus of the long arm are required for the formation of specific double- and triple-stranded coiled-coil structures. J. Biol. Chem. 1994, 269, 19167–19175. [Google Scholar] [CrossRef]
- Castiglia, D.; Posteraro, P.; Spirito, F.; Pinola, M.; Angelo, C.; Puddu, P.; Meneguzzi, G.; Zambruno, G. Novel mutations in the LAMC2 gene in non-Herlitz junctional epidermolysis bullosa: Effects on laminin-5 assembly, secretion, and deposition. J. Investig. Dermatol. 2001, 117, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Cserhalmi-Friedman, P.B.; Tang, Y.; Adler, A.; Krey, L.; Grifo, J.A.; Christiano, A.M. Preimplantation genetic diagnosis in two families at risk for recurrence of Herlitz junctional epidermolysis bullosa. Exp. Dermatol. 2000, 9, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Spirito, F.; Chavanas, S.; Prost-Squarcioni, C.; Pulkkinen, L.; Fraitag, S.; Bodemer, C.; Ortonne, J.P.; Meneguzzi, G. Reduced expression of the epithelial adhesion ligand laminin 5 in the skin causes intradermal tissue separation. J. Biol. Chem. 2001, 276, 18828–18835. [Google Scholar] [CrossRef] [Green Version]
- Chavanas, S.; Gache, Y.; Vailly, J.; Kanitakis, J.; Pulkkinen, L.; Uitto, J.; Ortonne, J.; Meneguzzi, G. Splicing modulation of integrin beta4 pre-mRNA carrying a branch point mutation underlies epidermolysis bullosa with pyloric atresia undergoing spontaneous amelioration with ageing. Hum. Mol. Genet. 1999, 8, 2097–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittwollen, R.; Wohlfart, S.; Park, J.; Grosch, E.; Has, C.; Hohenester, E.; Schneider, H.; Hammersen, J. Aberrant splicing as potential modifier of the phenotype of junctional epidermolysis bullosa. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Fine, J.D.; Mellerio, J.E. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: Part I. Epithelial associated tissues. J. Am. Acad. Dermatol. 2009, 61, 367–384. [Google Scholar] [CrossRef]
- Kiritsi, D.; Kern, J.S.; Schumann, H.; Kohlhase, J.; Has, C.; Bruckner-Tuderman, L. Molecular mechanisms of phenotypic variability in junctional epidermolysis bullosa. J. Med. Genet. 2011, 48, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Nakano, A.; Pulkkinen, L.; Murrell, D.; Rico, J.; Lucky, A.W.; Garzon, M.; Stevens, C.A.; Robertson, S.; Pfendner, E.; Uitto, J. Epidermolysis bullosa with congenital pyloric atresia: Novel mutations in the β 4 integrin gene (ITGB4) and genotype/phenotype correlations. Pediatric Res. 2001, 49, 618–626. [Google Scholar] [CrossRef] [Green Version]
- Klama-Baryła, A.; Rojczyk, E.; Kitala, D.; Łabuś, W.; Smętek, W.; Wilemska-Kucharzewska, K.; Kucharzewski, M. Preparation of placental tissue transplants and their application in skin wound healing and chosen skin bullous diseases - Stevens-Johnson syndrome and toxic epidermal necrolysis treatment. Int. Wound J. 2020, 17, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Cazzell, S.; Vayser, D.; Reyzelman, A.M.; Dosluoglu, H.; Tovmassian, G. EpiFix VLU Study Group. A multicenter randomised controlled trial evaluating the efficacy of dehydrated human amnion/chorion membrane (EpiFix®) allograft for the treatment of venous leg ulcers. Int. Wound J. 2018, 15, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Jin, R.; Powers, C.; Billi, A.; Bass, K. Dehydrated Human Amnion Chorion Membrane as Treatment for Pediatric Burns. Adv. Wound Care (New Rochelle) 2020, 9, 602–611. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, S.S.; Hall, L.; Chodosh, J.; Saeed, H.N. Long-term outcomes of amniotic membrane treatment in acute Stevens-Johnson syndrome/toxic epidermal necrolysis. Ocul. Surf. 2020, 18, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M.; Naderan, M. Management Strategies of Ocular Chemical Burns: Current Perspectives. Clin. Ophthalmol. 2020, 14, 2687–2699. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Jones, S.M.; Espinosa, M.; Green, V.; Nischal, K.K. Amniotic membrane transplantation in children with symblepharon and massive pannus. Arch. Ophthalmol. 2006, 124, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Altan-Yaycioglu, R.; Akova, Y.A.; Oto, S. Amniotic membrane transplantation for treatment of symblepharon in a patient with recessive dystrophic epidermolysis bullosa. Cornea 2006, 25, 971–973. [Google Scholar] [CrossRef]
- Hasegawa, T.; Mizoguchi, M.; Haruna, K.; Mizuno, Y.; Muramatsu, S.; Suga, Y.; Ogawa, H.; Ikeda, S. Amnia for intractable skin ulcers with recessive dystrophic epidermolysis bullosa: Report of three cases. J. Dermatol. 2007, 34, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Lo, V.; Lara-Corrales, I.; Stuparich, A.; Pope, E. Amniotic membrane grafting in patients with epidermolysis bullosa with chronic wounds. J. Am. Acad. Dermatol. 2010, 62, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, S.; Ramappa, M.; Mishra, D. Long-term outcome after superficial keratectomy in an infant with epidermolysis bullosa. J. AAPOS 2016, 20, 276–278. [Google Scholar] [CrossRef]
- Koulisis, N.; Moysidis, S.N.; Siegel, L.M.; Song, J.C. Long-Term Follow-Up of Amniotic Membrane Graft for the Treatment of Symblepharon in a Patient With Recessive Dystrophic Epidermolysis Bullosa. Cornea 2016, 35, 1242–1244. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Li, W.; Ling, S.; Sheha, H.; Qiu, W.; Li, C.; Liu, Z. Amniotic membrane extraction solution for ocular chemical burns. Clin. Exp. Ophthalmol. 2009, 37, 855–863. [Google Scholar] [CrossRef]
- Shayan Asl, N.; Nejat, F.; Mohammadi, P.; Nekoukar, A.; Hesam, S.; Ebrahimi, M.; Jadidi, K. Amniotic Membrane Extract Eye Drop Promotes Limbal Stem Cell Proliferation and Corneal Epithelium Healing. Cell J. 2019, 20, 459–468. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castiglia, D.; Fortugno, P.; Condorelli, A.G.; Barresi, S.; De Luca, N.; Pizzi, S.; Neri, I.; Graziano, C.; Trojan, D.; Ponzin, D.; et al. A Novel Phenotype of Junctional Epidermolysis Bullosa with Transient Skin Fragility and Predominant Ocular Involvement Responsive to Human Amniotic Membrane Eyedrops. Genes 2021, 12, 716. https://doi.org/10.3390/genes12050716
Castiglia D, Fortugno P, Condorelli AG, Barresi S, De Luca N, Pizzi S, Neri I, Graziano C, Trojan D, Ponzin D, et al. A Novel Phenotype of Junctional Epidermolysis Bullosa with Transient Skin Fragility and Predominant Ocular Involvement Responsive to Human Amniotic Membrane Eyedrops. Genes. 2021; 12(5):716. https://doi.org/10.3390/genes12050716
Chicago/Turabian StyleCastiglia, Daniele, Paola Fortugno, Angelo Giuseppe Condorelli, Sabina Barresi, Naomi De Luca, Simone Pizzi, Iria Neri, Claudio Graziano, Diletta Trojan, Diego Ponzin, and et al. 2021. "A Novel Phenotype of Junctional Epidermolysis Bullosa with Transient Skin Fragility and Predominant Ocular Involvement Responsive to Human Amniotic Membrane Eyedrops" Genes 12, no. 5: 716. https://doi.org/10.3390/genes12050716